
Background
CITRUS is an algorithm developed to automatically find stratifying signatures from within a data set to explain differences between multiple groups of samples. This article overviews the considerations and strategies for setting up and running an analysis. Click the links in the table of contents to jump to a particular section of this article.
- Create a New CITRUS analysis
- Selecting a Population
- Selecting Clustering Channels
- Assigning Sample Groups
- Compensation
- Scaling
- Association Models
- Cluster Characterization
- Event Sampling
- Minimum Cluster Size
- Cross Validation Folds (CVF)
- False Discovery Rate (FDR)
- Normalize Scales
- Plot Theme
- Analyzing results
- Failed or Defunct CITRUS Analysis
From within an Experiment, use the navigation bar, the experiment summary page, or an existing CITRUS analysis (copy settings) to create a new analysis.
Choose a population from the list of populations to determine which events are passed to CITRUS for analysis. The populations available for CITRUS are based on the Experiment Gates. Only one population can be chosen per analysis. If the population has tailored gates, events will be pulled appropriately based on the tailored locations. The typical strategy for choosing a population is to do some basic gating to eliminate dead cells, doublets, debris, beads, and other unwanted base populations. A typical example population for CITRUS could be CD45+ cells. Run CITRUS from this high level cleaned up population in order to maximize the discovery potential. Alternatively, downstream populations (e.g. CD3+ T cells) can be chosen instead to narrow the search space in cases where leads already exist.
The first step in CITRUS involves hierarchical clustering to divide the unstructured data into segmented populations. This step could be thought of as replacing the gating component of a typical analysis workflow, with the caveat that clustering does not assign named phenotypes. Named phenotypes are decided later by the investigator based on the the properties of the clusters.
The channels chosen for clustering are what provide the information by which data are subdivided into different populations. If a channel can be used to establish a phenotype, and would normally be used for this purpose with e.g. manual gating, then it should likely be included in clustering channel choices.
A channel should not be included as a clustering channel if the statistic choice for CITRUS analysis is medians and the channel will also be used for analysis (as is typical with, e.g., phospho-specific signaling markers). If the statistic choice for CITRUS analysis is abundances, a channel that will be used for analysis may also be included for clustering. More details on medians versus abundance can be found in the cluster characterization section below.
Missing channels for CITRUS? Note: the channels available to cluster on for a CITRUS analysis must be common to all files in the experiment. If two files have different panels, only those channels common to both will be available for CITRUS. The channel names must match by both their short and long name.
The core functionality of CITRUS is establishing biological explanations for why samples between two or more groups differ from each other. As such, indicating which samples belong to which groups is a necessary setup step, and this configuration is accomplished with the file grouping tool.
The minimum number of groups is two in order to run CITRUS. There is no maximum number of groups, but certain association models may enforce a maximum (e.g. LASSO currently has a maximum of two groups). Depending on the association model used, results may differ depending on the number of groups being analyzed simultaneously.
The minimum number of samples per group is three. However, for more robust statistical analysis and to avoid spurious results, at least eight samples are recommended per group.
If you already have assigned Sample Tags to your files, you can easily use them to filter the files and to assign them to a group. Simply click on the relevant sample tag in the Sample tags column: this action will automatically filter all the files for that specific sample tag. Then click on the group you want the filtered files to belong to and click on the Assign all displayed files option.

Importantly, if the files being grouped have useful information in their filename or sample name metadata, even if you haven’t assigned Sample Tags, you can use those terms during group creation to name the groups. The platform will scan the file names for the terms used in the groups and if the terms are found, the files will be automatically categorized.
Note: Currently, custom keywords within the FCS file that don't appear within the file name or the sample name are not accessible for searching to assign groups.
Compensation should be applied to fluorescent data before running CITRUS as it would be for any other analysis of these data, by selecting the appropriate compensation. The Cytobank platform uses the experiment-wide compensation to govern how compensation is applied to Gates, Illustrations and Advanced analyses. For files uploaded by DROP or FCS files that have no internal compensation matrix, you can leave the default option (file-internal compensation) and no compensation will be applied.
For a CITRUS run, the experiment-wide compensation will be used. If you wish to use another compensation, you can change the experiment-wide compensation on the Compensation page. Please be mindful when you do that since the change of experiment-wide compensation also affects your previously made gates and illustrations. Please see How the experiment-wide compensation works in Cytobank for more information.
When the experiment-wide compensation doesn’t match the compensation used for gating, there will be a warning message informing you to either update the experiment-wide compensation or redraw the gates to match the experiment-wide compensation before you are able to set up the CITRUS run.
All gates must be drawn on the same compensation in order to run CITRUS. In some rare cases where gates were drawn on different compensations, you will also see a warning message when you try to set up the CITRUS analysis. Please use the same compensation for all your gates before you proceed to a CITRUS analysis.
It is essential to set up the scales correctly before the runs. Scales affect CITRUS results because CITRUS analyzes data in scaled format. If uncertain about the quality of scaling in the experiment, validate that scaling looks appropriate ahead of time to avoid artifacts in the analysis. CITRUS is not affected by the minimum or maximum setting of the scales. The only thing that matters is the scale argument in the case of arcsinh scales. Please refer to the scaling support article and the blog post on how to scale cytometry data effectively for more details.
The selection(s) for association model determine the statistical methods used to discover stratifying signatures from clustered data features that explain differences between sample groups. There are three options:
- Significance Analysis of Microarrays (SAM) – Correlative
- Nearest Shrunken Centroid (PAMR) – Predictive
- L1-Penalized Regression (LASSO/GLMNET) – Predictive
The optimal association model to choose for any given case may depend on a variety of factors, and in general, the association models will often return similar results. Users unfamiliar with the association model options may elect to choose all three association models and compare the results. Users looking to build a model that can be used as a predictor of future data should use a predictive method. SAM is a correlative method and cannot be used in a predictive context. CITRUS analyses with more than two groups will have to use SAM or PAM, since currently LASSO does not support more than two groups. If false discovery rate is a desired metric within the results, PAM or SAM will have to be used since LASSO does not incorporate false discovery rate.
PAMR refers to the R implementation of PAM and may be referred to by either name. The mention of glmnet alongside LASSO references the glmnet package implementation of the LASSO method.
In single cell biology there are two principle ways to analyze and quantify individual samples. One approach is to identify cellular populations and subsequently quantify gene/protein expression (e.g. by median). Another approach is to quantify the abundance of various cellular populations within a sample. If expression levels of certain genes/proteins or relative abundances of cellular populations are different between sample groups, these differences can be used to establish a biological explanation for differences between sample groups or to identify biomarkers that are predictive of the sample group (e.g. disease group or treatment response) that a new independent sample belongs to.
CITRUS accordingly provides two modes for analysis of data in order to find stratifying biological signatures between sample groups:
1) Medians
In Medians mode, CITRUS will use the median expression levels of user-chosen markers in each cluster as features to find stratifying signatures between sample groups. After choosing medians, statistics channels from within the dataset will need to be chosen. Choose any channel(s) that you hypothesize might have differential expression between samples in specific cellular populations. Channels that were selected for clustering should not be selected again for statistics.
An example result could be that a particular subpopulation of monocytes has elevated levels of expression of a particular signalling marker in a group of non-reponders to a particular therapy as compared to the responder group. This difference in protein expression in this subpopulation of monocytes is indicative of a responder versus a non-responder to this therapy and can be used subsequently to predict future patient response to the therapy.
2) Abundance
In Abundance mode, CITRUS will use the relative abundance (proportion) of individual clusters in samples to find stratifying signatures of cell population differences between sample groups. The abundance proportion value is given by the number of events in the cluster divided by the number of events in the sample.
An example result could be that a group of non-responders to a particular therapy has a five-fold increase in a particular T cell subpopulation as compared to the responder group. This difference in the abundance of this T cell subpopulation is indicative of a responder versus a non-responder to a therapy and can be used subsequently to predict patient response.
Note: medians and abundance mode cannot be simultaneously selected. Get in touch with Cytobank Support if this would help your workflow.
There are two event sampling options for CITRUS:
1) Equal
In equal mode, each sample in the experiment will contribute the same number of events to the CITRUS analysis. The number of events taken per sample is capped at the number of events in the least abundant sample. Note that the number of available events per sample will also be dependent on the selected population.
2) Max per file
In max per file mode, each sample in the experiment can contribute a variable number of events to the CITRUS analysis. The value that can be entered in this field is capped at the number of events in the most abundant file. Note that the number of available events per sample will also depend on the selected population. Samples that do not have enough events to meet the specified event count will contribute all of their events.
Events sampled per file
This setting mediates how many events are taken from each sample for the CITRUS analysis. The cap on this value will depend on the chosen event sampling method and the selected population (see explanations for equal and max per file mode directly above). When a value is entered in this field, the total number of events to be sampled will be calculated below as Est. total events to be clustered.
The setting for minimum cluster size is similar to the setting for number of nodes in a SPADE run. Modulating this setting has the effect of lowering or raising the number of clusters in the final tree. A smaller number will result in more clusters. An important concept to remember when setting this value is that CITRUS removes from the analysis and ignores any clusters with a number of events that is smaller than the minimum cluster size. This strategy is taken to improve the statistical power of the modeling, since a larger number of clusters increases the number of statistical tests and therefore reduces the power of the model. Learn more about the removal of nodes from the CITRUS tree.
A useful guideline for setting the minimum cluster size is to use prior knowledge to evaluate the need for less abundant populations to be included in the analysis. Consider a CITRUS run with a minimum cluster size of 3%. If there is a dataset that contains a cell population that exists at a frequency of 1%, it will be removed from the analysis because it is below the minimum cluster size. In such a situation, the minimum cluster size should be decreased to accommodate the less abundant population.


( click to expand - the same dataset is clustered with a minimum cluster size of 5% (left) and 2.2% (right) )
The setting for cross validation folds (CVF) applies only to the predictive models (PAM, LASSO) and not to SAM, which is a correlative method. Cross validation is used in predictive modeling as a way to pick the parameters that will result in the most accurate model when the model is applied to new data. In CITRUS, the cross validation process is used to select the regularization threshold, which controls the number of features in the model.
Cross validation works by randomly partitioning the a complete dataset that is passed into the algorithm into multiple subsets (where the number of subsets is set by the CVF). All but one of these subsets are combined into a group (aka a "training set") in order to build the model with a particular set of parameters (in CITRUS, with a particular regularization threshold), and one subset (aka a "testing set") is used to test the model. This process is repeated with each subset being used as the testing set once for each parameter setting of interest. Then, the results from all of the samples when they were in the testing set are used to calculate the cross validation error for each parameter setting of interest.
The results of cross validation are an important component to interpreting CITRUS results, and are viewed on the CITRUS model error rate graph.
Two operational modes of CITRUS dependent on CVF setting
There are two principal modes of operation that CITRUS uses concerning cross validation. They lead to different behavior of the algorithm which is not apparent from the setting alone. The key components of the different modes of operation are the clustering and model building steps. The different modes are explained below:
Case of CVF equal to 1
A cross validation fold setting of 1 engages CITRUS in a special operational mode that can noticeably increase the speed of the run but may bias the results and therefore diminish the applicability of generated models to other independent datasets. When the CVF is set to 1, CITRUS clusters all of the data together and then applies a cross validation procedure only for the model building (this limited cross validation is done with 10 folds or the maximum number possible given the number of samples and groups). Since the data are clustered only once, the estimates of accuracy given by the cross validation error rate may be overly optimistic.
Case of CVF greater than 1
A cross validation fold setting greater than one performs cross validation at the number of folds specified on the entire process of clustering and model building. That means that the samples are first divided into as many cross validation fold subsets as are specified in the CVF setting for the run. After this, the clustering, model building, and testing occur within every cross validation fold as explained above. Since, in any given cross validation fold, the samples in the testing set are not clustered along with the samples of the training set, a nearest-neighbor mapping routine is used to map the testing set samples onto the clusters of the training set. This is similar to what would be required if new data were to be used in a model, and is therefore important for estimating accuracy in a model that would be generalizable to new samples.
Currently in Cytobank any CVF below 5 that is not equal to 1 is not permitted. This is because CVFs between 2 and 4 do not result in robust cross validation.
If the CITRUS run is exploratory or if you want a quick look at the data to see if there is any signal from stratifying signatures, CVF = 1 may be a good place to start. Using a CVF greater than 1 is recommended for publication-quality results (10 is a common value used in statistical applications).
There are different costs and benefits associated with choosing a cross validation of 1 or greater than 1. They are discussed below:
1) Cross validation thoroughness
A misconception when using a CVF of 1 is that it implies a lack of cross validation (or, equivalently, a single fold of cross validation). As discussed above, this is actually not the case in CITRUS. When CVF is set to 1, a 10 fold cross validation the modeling step is still performed. However, unlike with a CVF setting greater than 1, the clustering is not included in the cross validation (see the implications of this below).
2) Algorithm run time
With CVF equal to 1, the dataset is only clustered once and those clustering results are used throughout the run. When CVF is set to larger numbers, the number of clustering steps increases according to the number of cross validation folds (discussed above). Clustering is the most time consuming part of a CITRUS run. Consequently, substantial increases in the overall execution time will be seen as the CVF is increased. Consider the results of this simple test of a CITRUS run with 16 files in 2 groups, clustering 140,000 events across 10 channels, and analyzing 14 signaling channels using PAM. The run was repeated three times with different CVF settings:
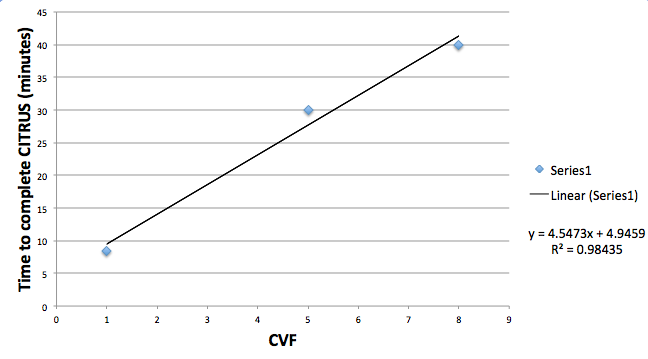
(CITRUS run time as a function of CVF setting for one particular dataset and configuration - results may vary)
3) Applicability of the model to other independent datasets
The decision to set the CVF greater than 1 will also hinge heavily on the degree to which the model is expected to generate results that will apply to independent datasets or broader contexts. When the CVF is greater than 1, the test set samples in each cross validation fold are mapped onto clusters that were built without them. This is similar to what would be required if new data were to be used in a model, and is therefore important for estimating accuracy in a model that would be generalizable to new samples. This is why it is recommended to use a CVF greater than 1 for publication-quality results.
When using features from a dataset to establish a model, as the number of features used increases, the chance that one or more of the result features is coincidental (a false positive) instead of a true association also increases. The FDR threshold helps to make sure that models produced by PAM and SAM don't include too many false positive features.
In the case of PAM, the user can enter the threshold for false discovery rate. Lowering the false discovery rate will limit the number of features that will be included in the model. Increasing the false discovery rate will do the opposite. This only applies to one of the multiple results packages that emerges from using the PAM association model, termed the fdr_constrained model. Learn more about CITRUS results constraint groups.
When the Normalize Scales option is turned on, each channel is normalized such that it has a mean value of zero and a standard deviation of 1. This is done by first concatenating all files within the CITRUS run, then for each event value per channel, subtracting the mean and dividing by the standard deviation of the channel. Normalizing scales can be a useful strategy when channels have different dynamic ranges, as is often the case in fluorescence flow cytometry.
The plot theme setting simply changes the background color of images and figures within the CITRUS results.
Learn how to analyze CITRUS results!
On occasion a CITRUS run may fail or enter into a defunct state.
Failed CITRUS run - The cause of CITRUS failures are often related to the dataset itself and can be identified and fixed without much effort. Get in contact with Cytobank Support for help with this process. If possible, please include the CITRUS log file that can be downloaded from the interface after the CITRUS failure.
Defunct CITRUS run - This issue arises from the CITRUS run consuming more compute resources than are currently allocated for it by Cytobank. In order to make the CITRUS run succeed, the right compromise of settings that result in less consumption of compute resource will have to be found with some degree of trial and error. The following adjustments can raise the chances of success for a CITRUS run that previously went defunct:
- Decreasing the number of events in the analysis.
- Decreasing the cross validation folds, especially to a value of 1, which dramatically decreases run time and resource use (learn more).
- Increasing the value for minimum cluster size.
- Decreasing the number of clustering channels.
Of these options, 1 and 2 will make the biggest difference. 3 and 4 make less of a difference and are not ideal because changing them might not be possible for the scientific question being asked.