
Background
Once CITRUS has been successfully run, it becomes important to know how to analyze the results. This article explains all the components of CITRUS results, how to interpret them, and all the details along the way.
Use the links below to jump to different sections of the article:
- Downloading the CITRUS Results Package
- Results Package - CITRUS Run Information File
- Results Package - Supporting Files
- Results - CITRUS Trees Colored by Channel
- Important Notes On CITRUS Tree Structure
- Redundant events
- Each parent cluster has two children and in some cases one child
- The numbers on the clusters are identifiers
- Association Model Results - PAM, SAM, LASSO
- Interpreting the Model Error Rate Graph
- Next Steps for CITRUS Results Analysis: Export Clusters
Downloading the CITRUS Results Package
By default, there are no interactive results for a CITRUS run. Unlike SPADE, there is no interactive tree viewer or results explorer. The results must be downloaded as a package from the setup page of the CITRUS run itself or from the experiment summary page within the CITRUS section.
![]()
(link to download CITRUS results after a run is completed on the setup page)
(link to download CITRUS results after a run is completed on the experiment summary page)
If an interactive tree viewer would make a difference in your CITRUS workflow, please get in contact with Cytobank Support to discuss how.
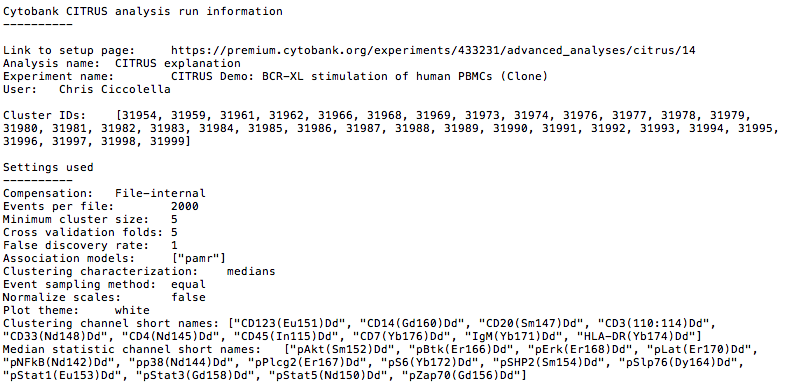
Results Package - CITRUS Run Information File
The top level of each results package for CITRUS is accompanied by an information text file. This file contains essential attributes of the run including basic metadata such as which user executed the run, what settings were used, the IDs of nodes from clustering, and URLs back to the setup page in Cytobank. This helps improve traceability of results packages that get downloaded off of Cytobank onto local file systems.
Results Package - Supporting Files
In the top level of the CITRUS results package there is a folder called supporting_files. This folder will generally not help interpret the results of the analysis, but contains important supporting files for other uses. For example, these files are used behind the scenes when writing new FCS file based on CITRUS clusters, and also can be used as a complete record of settings at the time of analysis. For users familiar with R, one of the files can be loaded in R to extract information that could be useful for other downstream applications.
citrusClustering.rData - This file can be loaded in R and contains information about the settings and details about the CITRUS analysis and the model that was built. It can be used in R for downstream analysis.
expt_run_files - This folder contains records of the channels and scale settings at the time of analysis.
r_script_bg_task... - This file is a log file of the CITRUS run.
Results - CITRUS Trees Colored by Channel
The first part of the CITRUS algorithm involves hierarchical clustering in order to group cells into populations. This clustering results in a tree that may look similar to that generated from SPADE. To view these trees, navigate into the results folder of the CITRUS results package and find the two PDFs called markerPlots.pdf and markerPlotsAll.pdf. The trees displayed in these PDFs are the same; the difference between the PDFs is the size of the figures: one contains a single page of smaller figures and the other contains multiple pages of larger figures.
The CITRUS tree is displayed multiple times, but each version of the tree is the same and represents all of the cells from all of the samples clustered together. Each time that the CITRUS tree is displayed, it is colored by different channels from the data in order to illustrate which clusters contain cells that are positive for certain markers. The color scale bar indicates the transformed intensity of marker expression per cluster according to the scale settings in the experiment. Note: no channels besides the clustering channels will be shown in the colored tree PDFs. Get in touch with Cytobank Support if this functionality would help your workflow.
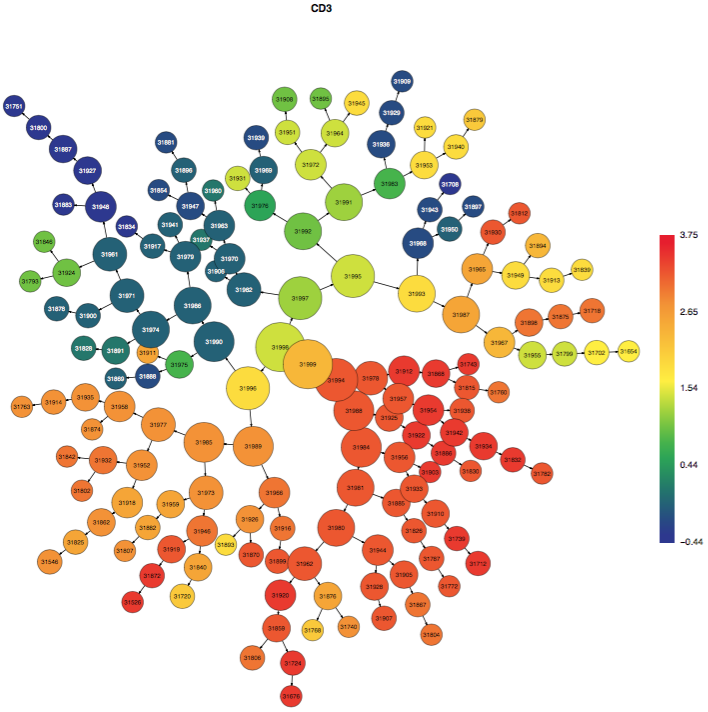
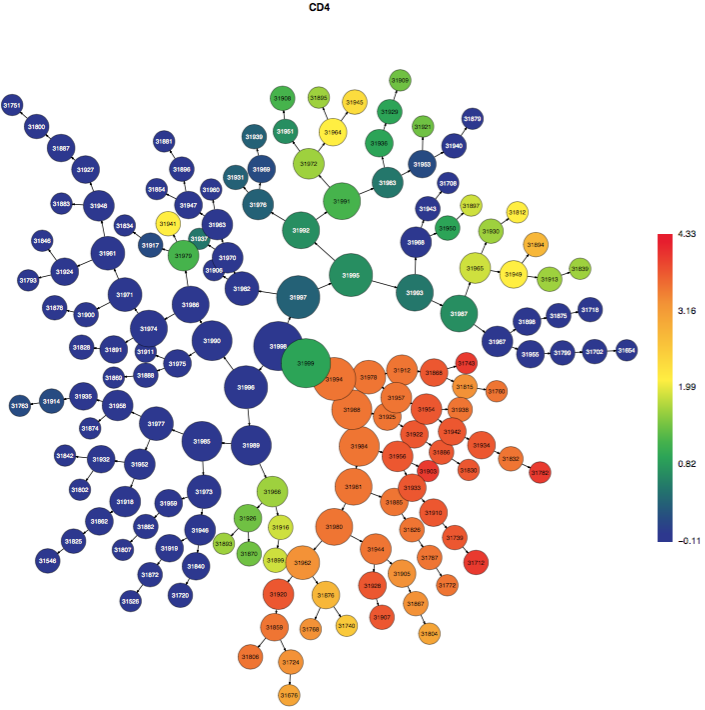
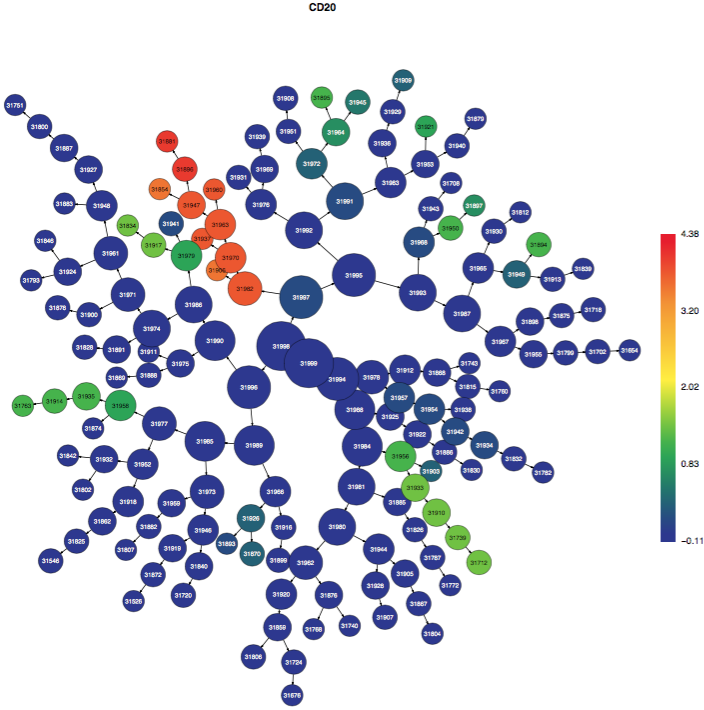
(click to expand - CITRUS trees colored by different channels - familiar populations emerge as branches)
Important Notes On CITRUS Tree Structure
There are some important concepts to understand when interpreting the CITRUS tree.
Unlike SPADE or other similar clustering algorithms, CITRUS represents events redundantly in the tree. Put another way, any given event can contribute to multiple clusters. CITRUS does this in order to make sure that the algorithm is finding the best clusters to optimize the prediction of the differences between the groups - it helps get around having to decide how many clusters there should be.
In order to understand this redundancy on the graph, remember that each parent cluster contains the events of its children. This pattern starts with the center cluster. It is the largest cluster, has no parent, and contains all the events in the analysis. The events in this parent cluster are then split into two child clusters and this process is repeated, each time with the parent retaining the events in the children. The events in a child node are represented in every connected node above them all the way to the origin of the tree. To help you understand the relationship of the clusters, the CITRUS trees are displayed with arrows between the clusters that always go from the parent to the child.
This concept of redundant events especially important to understand when exporting clusters for downstream analysis so as not to create unnecessary complexity and extra work in these downstream analyses. For example, consider a discovery such as the one below. There are three nodes that each have a stratifying signature that predicts membership of a sample in the ref group or the bcr group. However, the three nodes contain some redundant events because of their sequential parent and child relationship. Exporting all three nodes for downstream analysis would create many events in triplicate in the resulting FCS files and may be confusing. A good recommendation in this scenario is to export only the parent node in this hierarchy (node number 31947), which will contain all of the events in both child clusters:
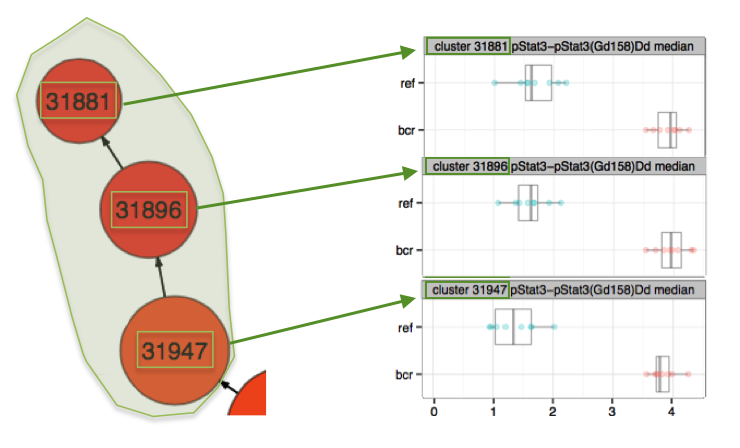
(keep in mind redundant events when interpreting data or exporting clusters for downstream analysis. The events in cluster 31881 are in cluster 31896 and all of those events are in cluster 31947)
2) Each parent cluster has no more than two children and in some cases only one child
By design, each cluster in CITRUS will be split into two child clusters until the most granular clustering is achieved (as determined by the user-specified Minimum Cluster Size for the run). As mentioned above, each parent cluster contains the events that are in its children and its children's children, and so on. The cells in each parent cluster are split into two child clusters that are displayed on the tree graph. As the tree progresses, however, CITRUS clusters with only one child are commonly displayed. Parent clusters with only one child displayed occur when one of the two child clusters that would have been displayed has a number of events (across all of the samples) smaller than the Minimum Cluster Size set before the run. In this case, where only one child of a parent is displayed, the child that is not displayed is also not used in the model building and analysis. Thus, the Minimum Cluster Size set by the user determines how granular the clustering gets.
Using a higher Minimum Cluster Size leads to a lower number of clusters which reduce the number of performed tests and improves the statistical power of the model. Since a large number of clusters would mean that the model has to do and correct for more statistical tests. However, if you are interested in rare cell populations and they are not shown at the end of your CITRUS trees, you may want to consider decreasing the Minimum Cluster Size.
3) The numbers on the clusters are identifiers
The number displayed on each cluster is an identifier. The identifier will show up in other places within the results and can be used to export the cluster to an FCS file for downstream analysis. The number displayed on the origin parent cluster corresponds to the number of events in the analysis minus 1.
Association Model Results - PAM, SAM, LASSO
The results reported from the CITRUS association models are the features that have statistically significant differences between samples in different groups for a given cluster. The results package includes information about the cluster, the features of the cluster that are significantly different between sample groups, and some supporting information to help you understand the difference and the phenotype of the cluster(s). Beyond this, clusters of interest can be exported as new FCS files within Cytobank for any type of downstream analysis that is desired.
Results folder organization
The key CITRUS results are found in a folder inside the results folder that is named according to which association model was chosen for the analysis. If multiple association models were chosen for a single run, multiple results folders will appear for each association model.
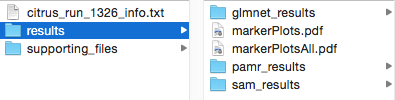
(CITRUS results for a run that had all three association models selected. Note that glmnet = LASSO)
Components of association model results
There are five total components of the association model results in a successful CITRUS run. They are:
- clusters...pdf - used for understanding phenotype of significant clusters. Each cluster phenotype is plotted as a histogram against the background phenotype. This visualization shows all the data files passed into CITRUS as concatenated together into one pool. The cluster histogram shows the part of the data that falls into the indicated cluster. The background histogram shows all the rest of the data that don't fall into the indicated cluster. By comparing the difference between the cluster phenotype and the background phenotype, the distinct phenotype of the cluster can be discerned.
- featurePlots...pdf - used as a visualization of the clusters in the tree that are part of the significant results. This visualization can be used to focus attention on particular areas of the tree before interpreting the plots of CITRUS trees colored by channel.
- features...csv - spreadsheet detailing samples and the relevant statistics for significant clusters. Note that median values for channels are transformed according to scale settings at the time the CITRUS run was started.
- features...pdf - graphs displaying the median expression or abundance differences between samples in significant clusters. In a CITRUS run with a cluster characterization setting of medians, each data point on these graphs represents the median intensity of a single marker for cells in a single cluster in a single sample. The scale of the graph corresponds to the scaling of the data (this will generally be transformed values). In a CITRUS run with a cluster characterization setting of abundance, each data point on these graphs represents the proportion of the event count of the cluster compared to the total event count in a single sample. Values are decimal equivalent of a percent between 0 and 1 (for example, a cluster with 100 cells in a sample with 1000 cells would have a value of 0.1). Note that the scale for abundance indicates "Log10 scale", but this is simply a labeling error. There is no log10 transformation of data; only the calculation done for proportion as described.
- ModelErrorRate.pdf - used for assessing the quality of results for predictive models only (PAM, LASSO). SAM does not have this graph because it is a correlative method, not a predictive method. See dedicated section below for more details.
Note: If CITRUS does not find any results then these components will not appear with the exception of the ModelErrorRate graph for PAM and LASSO.
Association model constraint groups
The ellipsis (...) found in the components listed above is present because each results component will appear multiple times according to which constraint group it belongs to. Each constraint group is a possible model result that could be used depending on the goals of the analysis. The constraint groups are generated as part of the evaluation of cross validation error rate (for PAM and LASSO) and false discovery rate (for all three statistical methods) that CITRUS does and that is visualized on the Model Error Rate Graph. As a result of this evaluation, CITRUS generates up to three results per association model, each a result of constraining the cross validation error rate and/or false discovery rate in some way.
The constraint groups vary between being more conservative with fewer defining features and less conservative with a greater number of defining features. The notations for the constraint group results and their details are described below for each statistical method.
PAM and LASSO
- cv_min - these results correspond to the model with the fewest number of features necessary to have the lowest cross validation (cv) error rate.
- cv_1se - these results correspond to the model with the fewest number of features necessary to have a cross validation error rate that is 1 standard error higher than the minimum. This value is commonly used in statistical applications to select a model that is most likely to be the best predictive model on new data.
- cv_fdr_constrained - (PAM only) these results correspond to the model with all of the features that can be included below the false discovery rate (fdr) set by the user during the setup process for this run (default = 1%). LASSO as implemented in CITRUS does not factor in false discovery rate.
SAM
- fdr_0_01, fdr_0_05, fdr_0_10 - each of these results corresponds to the model with all of the features that can be included below a false discovery rate (fdr) of 1%, 5%, and 10%, respectively. SAM does not have cross validation step because it is a correlative method, not a predictive method.
The notations of cv and fdr within the results stand for cross validation and false discovery rate. Cross validation is the process by which the accuracy of a predictive model is estimated. False discovery rate is a metric designed to minimize the chance that the discovery being made (i.e. the feature being reported as significantly different between groups) is actually coincidental (i.e. a false positive).
Interpreting the Model Error Rate Graph
The model error rate graph accompanies the results for CITRUS runs that use the predictive methods (PAM and LASSO).
Testing multiple models with different numbers of features
The first key to understanding the model error rate graph is to consider that CITRUS creates and evaluates multiple models in order to find the best one for its predictive analysis. Each model is bound by different regularization threshold that in turn impacts the number of features in the model. A feature is a cluster and a trait of that cluster. For any given CITRUS run, a trait will either be the relative abundance of events in that cluster, or the median intensity of a biological marker in that cluster, depending on the setup parameters of the run. Each data point on the model error rate graph is a model that CITRUS is evaluating. The number of features in each model is shown at the top of the graph.
Cross validation error rate
For the predictive association methods (PAM and LASSO), each model CITRUS is evaluating has to be judged for its prediction accuracy.
This is done using a common statistical practice called cross validation, which randomly partitions the a complete dataset into multiple subsets. Repeatedly, all but one of these subsets are combined into a group (aka a "training set") in order to build the model with the regularization threshold that CITRUS is evaluating, and one subset (aka a "testing set") is used to test the model. The results from all of the samples when they were in the testing set are used to calculate the cross validation error rate for the regularization threshold CITRUS is evaluating.
The cross validation error rate is the percentage of times that the model is wrong at predicting the sample group that a file belongs to when it is not included in the training set to build a model. In order to understand the cross validation error rate, it may help to keep default expectations in mind. If you had zero features in the model, the cross validation error rate would be the result of a random guess. In a CITRUS run for two sample groups with an equal number of samples per group, the default expectation for the cross validation error rate should be about 50%: equivalent to a random guess for the group that each sample belongs to. For three groups, the default expectation should be around 66%.
Note: CITRUS does not account for groups with unequal numbers of samples in calculating the cross validation error rate. If your sample groups are unbalanced, it is important to understand this when evaluating the cross validation error rate. For example, consider a CITRUS run with a total of 100 samples and with 90 samples in group A and 10 samples in group B. If the cross validation error rate is 10%, it may be because the model is simply predicting that all of the samples are from group A (therefore getting the prediction of all 10 samples from group B wrong). However, a 10% cross validation error rate could also result from the model calling 5 samples from group A incorrectly and 5 samples from group B incorrectly. Therefore, when the sample groups are unbalanced, it is difficult to make a straightforward interpretation of the cross validation error rate.
False discovery rate
When using features from a dataset to establish a model, as the number of features used increases, the chance that one or more of the features is a coincidental result (false positive) instead of a true association also increases. The false discovery rate metric is used as a way to increase the confidence of the results in models produced by PAM and SAM.
Next Steps for CITRUS Results Analysis: Export Clusters
A successful CITRUS run will produce results that indicate that features of certain clusters of cell populations within the data are significantly different between groups of samples and may be important to understanding the underlying differences in these sample groups (e.g. responders versus non-responders). The CITRUS results package provides basic information to understand the cell phenotype that corresponds to the significant clusters and other data about these clusters, but most investigators will want greater freedom to explore and analyze the events that CITRUS has isolated in other ways. In order to accommodate this, Cytobank supports the export of CITRUS clusters as new FCS files for any desired downstream analysis.