
Table of Contents
This article overviews DROP, a functionality that accepts any numerical spreadsheet file for analysis within Cytobank.
DROP is currently available on Enterprise Cytobank with full functionality and Premium Cytobank with some limitations.
Click the links below to jump to the relevant article section.
- Background
- Acceptable Data Inputs and Formatting Requirements
- How to Use DROP in Cytobank
- Analytical Strategy Guidance
The investigation of biological systems will often encompass the use of numerous technologies to understand states and processes in different compartments of organisms and their cells. Many of these technologies produce data in a format that is generally the same as that found in flow or mass cytometry data, i.e., a numerical matrix with rows representing observations (traditionally single cells) and columns representing the relative quantity of a biological marker being measured (traditionally proteins). Due to this similarity, many of the tools and workflows for manual and automated analysis of data with Cytobank that have been predominantly used for cytometry are also useful for the analysis of other data, such as that derived from DNA, RNA, imaging, clinical information, other protein measurements, metabolomics, etc, and whether produced from single cells or bulk cell mixtures. DROP is able to import any data in the format of a spreadsheet / numeric matrix for analysis by converting it into an FCS file within Cytobank.
Acceptable Data Inputs and Formatting Requirements
- DROP accepts data in comma-separated (CSV), semicolon-separated, or tab-separated (TSV) spreadsheet format of rows and columns where all data points are numeric.
- If non-numeric data points are present and useful for analysis, consider replacing them with integers to code the different values. Then use category/cluster gates to isolate the relevant data points in Cytobank.
- There is currently a maximum of 818 columns permitted for a DROP conversion on the Enterprise Cytobank and 100 columns for Premium accounts. For datasets with larger numbers of columns than this, pare down the number of columns by filtering on variance or by running PCA.
- Various header information is permitted in the input file above the actual data, but the first row of data must be names for each column of data. The names will subsequently be displayed as FCS channel names in Cytobank.
- Names will be copied to the short and long channel name of the converted FCS file. In order to specify a unique short and long name, separate the two inputs with a double underscore. For example, "shortname__longname". Learn more about channel names.
- If converting multiple files at the same time, all files must have the same row/column data start point.
- The number of data points per row must be consistent throughout the entire input matrix.
- Duplicate column names are not permitted.
This is a step-by-step guide for how to use DROP in Cytobank. To try it out, download the dataset attached at the bottom of this article and follow along.
1) Enter an Experiment
Create a new Experiment or access an existing Experiment in Cytobank.
2) Enter the Upload Files page
This page will be mandatory if creating a new Experiment. Otherwise it can be accessed in an existing Experiment.
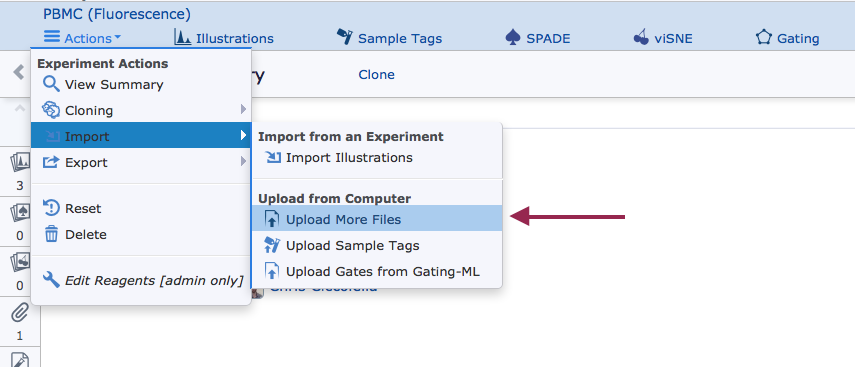{kind=link}
3) Select Files for Conversion
Use drag-and-drop or the selector button to choose files from the computer to upload and convert with DROP. Note that normal FCS files can also be uploaded at the same time. Cytobank will identify files that are able to be converted and present the option to convert them. If the box for file conversion is deselected, the files will simply be uploaded as attachments.
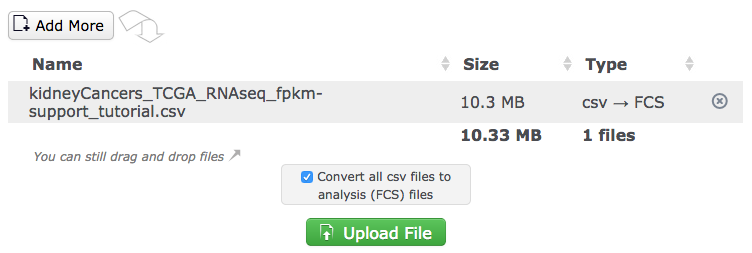
(Files are displayed after being dragged into the receiving area. Clicking the Upload File button will initiate upload and steps for configuring conversion)
Note that due to column limits, datasets may need to have their set of features reduced before upload to DROP. Useful strategies for this include filtering by variance. Also note that data may need to be transposed if data labels are at the beginning of each row instead of each column. This cannot be done by Cytobank.
4) Configure the Conversion
Click the upload button to begin the upload. It will proceed in the background. Before the conversion can happen, however, it must be configured. First answer the prompt about which data type is being converted in order to inform Cytobank product development efforts. Next, use the interactive data snapshot prompt to mark the beginning point of the numeric data matrix. Note that the column names will be implied from the row right above the numeric start point specified.
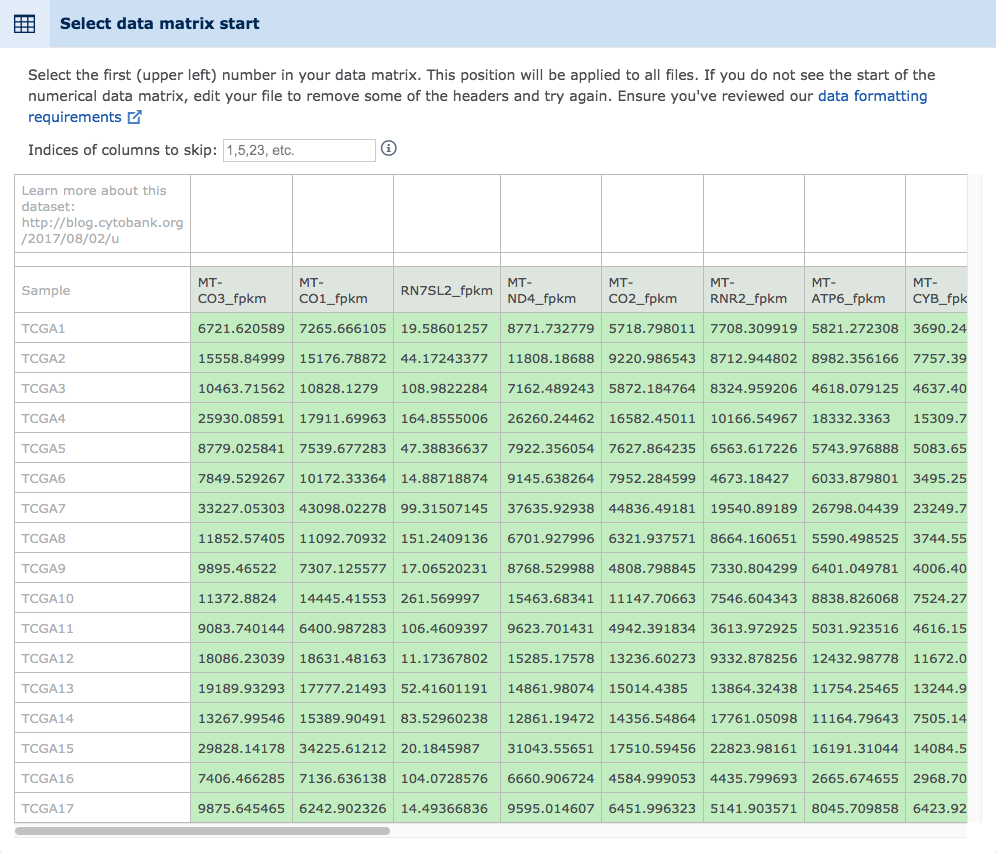
(Selecting the data matrix start point with the mouse. The numeric start point should be selected and the column names will be assigned automatically. Header information and non-numeric beginning columns can be skipped. The entry box above the data matrix can be used to specify columns from within the data to skip)
5) Review the Conversion Summary
Each successful conversion will display a summary box. This information should be reviewed for any warning messages or useful information associated with a successful transfer and to verify that the conversion matches expectations. Failed conversions will display a different screen with error messages that can help in addressing problems.
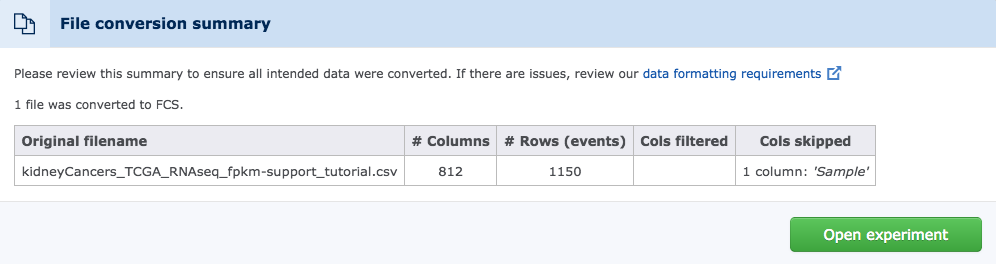
(The conversion summary offers useful information and potential warnings about the conversion)
Click the Open Experiment button to proceed with analysis as normal in Cytobank.
For information about how to use DROP via the Cytobank API, check out the API Documentation and programming libraries.
General Notes
The analytical strategies taken on any converted data will depend on the nature of the data itself. In particular, the algorithms offered within Cytobank are not always applicable to all data types. In order to decide what analytical methods are appropriate, evaluate the type of data against the table below either by the example row or from first principles based on the data properties.
| Example Technology | Data Properties | Plots / Stats | viSNE | SPADE | FlowSOM | CITRUS |
| Cytometry, RNA-Seq, segmented image features | Single Cell Continuous | Yes | Yes | Yes | Yes | Yes |
| RNA-Seq, Protein expression, clinical features, cell population features | Bulk Continuous | Yes | Yes | Yes | Yes | |
| bulk DNA, clinical features | Bulk Ordinal | Yes | Yes |
(Table for mapping data types to analysis options. "Continuous" describes data that represents a quantity or magnitude measurement of some trait of the observation. For example, the relative quantity of some gene or protein. "Ordinal" describes data that take the form of discrete categories that have a natural order. For example, pain on a scale of 1-10 or genotype at a locus (0,1,2). Categorical data that do not have a natural order are termed "nominal" and shouldn't be used in any algorithmic analysis, though they can be used for useful visual annotations if they accompany continuous or ordinal data)