
Background
After successfully setting up and running a dimensionality reduction analysis, you can use your viSNE/opt-SNE/tSNE-CUDA/UMAP map for exploratory data analysis and for visualization of the results of other, more quantitative downstream analyses. If you have run more than one dimensionality reduction algorithm you can also compare results with other dimensionality reduction algorithms. In this article, we refer to all four dimensionality reduction algorithms (viSNE/opt-SNE/tSNE-CUDA/UMAP) as DR.
Before doing this, you’ll want to do a quick check of the DR map quality to see whether you want to kick off another run after tuning any of the advanced dimensionality reduction settings. This article outlines methods and considerations for assessing DR map quality, doing exploratory analysis with DR algorithms, and using them for visualization of downstream analyses. Click the links below to jump to the relevant section:
Assess DR quality
- Color the DR map by clustering channel
- Assess DR map quality
- Assess viSNE map quality
- Assess opt-SNE/tSNE-CUDA map quality
- Assess UMAP map quality
Exploratory data analysis with DR
- Color the DR map by functional markers not used for clustering
- Assess differences between groups or quality control with DR contour plots
- Visualize sample heterogeneity across multiple dimensional with DR grid
- Color the DR map by additional overlaid variables: manual gates, disease groups, etc.
- Concatenate DR map across samples
Downstream analyses using DR for visualization
- Gate the DR map to define populations or groups manually
- Create a heatmap of channel expression for all manually gated DR populations
- Use clustering to automatically define populations or groups and display them on the DR map
Assess DR quality
Color the DR map by clustering channel
A key workflow for assessing the quality of a DR map is to color it by channel. Use the dot plots colored by channel functionality to color each event in the DR map according to its intensity on a channel within the dataset. The patterns that emerge show why dots in the map are nearby each other, or which markers make them similar to each other. In cell phenotyping, you'll see events close together on the DR map based on their phenotypes. In bulk sample heterogeneity analyses, you'll see samples close together on the DR map based on their similarity across multiple biomarkers. One good way to set up the Illustration to quickly and easily assess your DR map quality is with the clustering channels on the columns and the files or a subset of the files on the rows, like in the example below. Setting up a view like this will allow you to quickly assess DR map quality, as explained in the section below.
(click to expand - dot plots colored by channel on viSNE map within the Illustration. The rows display selected files, the columns are channels, the color intensity bar on the right side of each plot represents the marker expression of each channel.)
(click to expand - dot plots colored by channel on UMAP within the Illustration. The rows display selected files, the columns are channels, the color intensity bar on the right side of each plot represents the marker expression of each channel.)
To see more instructions on Illustration settings, please click here.
Before proceeding with exploratory data analysis or visualization of downstream results on a DR map, you’ll need to assess whether the DR settings used in your run produced a good quality map. Even with good quality data, you might get a poorly resolved DR map if you haven’t used the optimal settings for your data type, panel, and number of events. When you color on the clustering channels as described above, a poorly resolved DR map will have overlapping and poorly formed islands that don’t separate the expression of a single marker into distinct locations on the map.
Assess viSNE map quality
This example compares a poorly converged viSNE map and a nicely converged viSNE map across several markers:
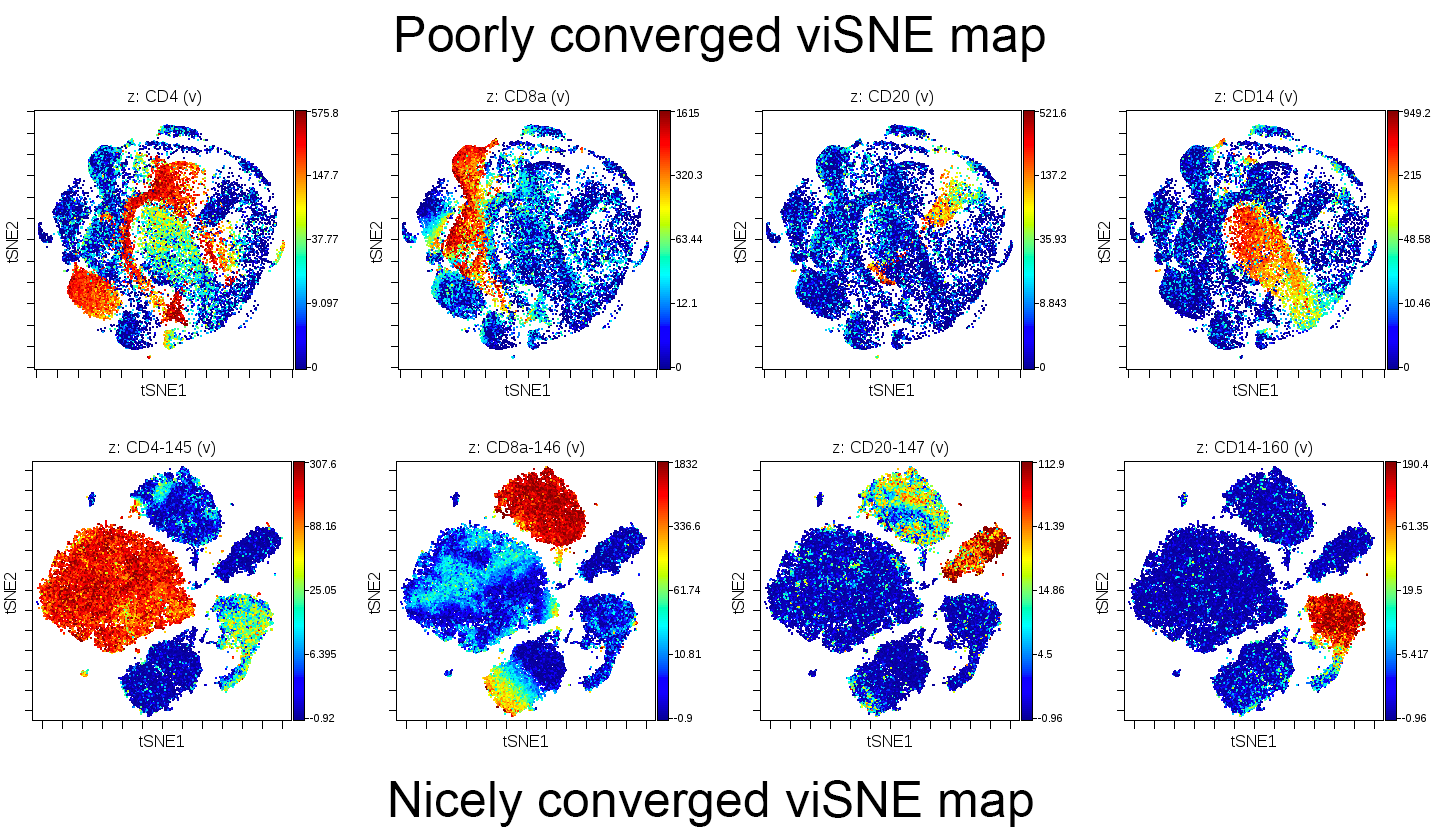
(viSNE maps colored by channel comparing a poorly converged viSNE map (top) to a nicely converged viSNE map (bottom). Each row is a single sample colored by four different markers as indicated. The two samples are not related and are from different viSNE analyses. They are shown together only for purposes of comparison and reference for the idea of poor versus good convergence.)
The most typical reason for poor convergence is a lack of iterations when working with viSNE runs with larger numbers of events (as a rough example, in abundance of 400,000 events). If coloring the viSNE map by channel results in overlapping and poorly formed islands of events that don't separate the expression of a single marker into distinct locations on the map, another run should be attempted with more iterations to improve the clarity of the results. In addition, changing the perplexity may improve the separation of events in the viSNE map.
Note that if you have pre-gated to a fairly granular starting population for your viSNE, such as CD4 T cells or B cells, you will generally not see distinct islands resolved within these pre-gated populations, but you should see cells with high expression of a single marker appearing in distinct locations on the map.
Assess opt-SNE/tSNE-CUDA map quality
The settings of iterations for tSNE-CUDA and max iterations for opt-SNE are also important to create optimized maps. The max iterations are the maximum number of iterations it will run. The actual number of iterations performed should be smaller than the max. Make sure to not set the opt-SNE max iterations too low so that the algorithm will stop prematurely before the necessary iterations to finish the embedding. You will be able to see the number of iterations performed at the completed opt-SNE analysis page. If the iteration performed is equal to the max iterations, which could be an indication of lack of iterations, try increasing the max iteration and run again.
The iterations for tSNE-CUDA can be set automatically. The formula is number of events/1500, with a minimum of 750 iterations as default. In most cases, the automatic iterations work well. If you wish to change the number of iterations, make sure not to set it too low. Lack of iterations will result in sub-optimal maps.
(Examples of opt-SNE and tSNE-CUDA map with different iteration settings, good map [left column] and poorly embedded map [right column])
Assess UMAP map quality
This example compares UMAP with different minimum distance (MD) settings. This value is the effective minimum distance between embedded points. Smaller values will result in a more clustered/clumped embedding where nearby points on the manifold are drawn closer together, while larger values will result in a more even dispersal of points (Nolet, Lafargue, Raff, Nanditale, Oates, Zedlewski, and Patterson, J, Preprint (2021)). In most cases the default value of 0.01 works pretty well. It’s a good idea to start with the default value and increase the value as needed based on the initial run.
(UMAP with different minimum distance values [MD] from the default 0.01 to the largest 0.99. CD3 marker is showing as the Z-channel to indicate the CD4 expression level. As the MD value increases, the embedding is more clumped, and if it goes too high, in the case of MD 0.5 and 0.99, the islands are merged and not able to separate the populations)
Exploratory data analysis with DR
Color DR map by functional markers not used for clustering
If your panel includes markers that you are interested in studying but were not included as clustering markers for the DR analysis (for example, signaling markers, activation markers, or inhibitory receptors), you can use dot plots colored by channel as described above for the clustering markers to color the cell populations or groups of samples on your DR map according to their expression of these markers. If you’ve already gated on the DR map or used a clustering method for automated categorization of the cell populations, you can also display the gate labels as you view functional marker expression for easier interpretation.
(viSNE maps colored by expression of p-NFkB, a functional marker that was not used to cluster the viSNE. p-NFkB cell subset gate is shown and labeled, facilitating the comparison of this subset across the three unique conditions - basal, BCR, and IL-7)
(here is another example showing a UMAP map colored by expression of p-NFkB, a functional marker that was not used to cluster the UMAP; p-NFkB cell subset is shown red at 10 o’clock across the three unique conditions - basal, BCR, and IL-7)
Note that this type of visual can also be used to visualize the correlation between continuous meta-data variables (e.g., age) and cell populations or groups of samples.
Assess differences between groups or quality control with DR contour plots
Oftentimes cell populations will be present at some level in multiple samples under different conditions or from different clinical groups, but the abundance of these populations will differ across samples. Contour plots are a great way to quickly visualize these differences in abundance. With single-cell data, you can use them to see differences in abundance across samples from different experimental or disease conditions (e.g., unstim vs stim or responder vs non-responder), or to locate any potential differences across batch if the same technical control sample is run across repeated experimental batches. To make this illustration, use the same settings as depicted above (e.g., make x and y axes tSNE1 and tSNE2 or other DR channels.) but change Plot type to Contour and Color by to Density. An example figure using viSNE contour plots is shown below, and you may use any other DR channels to have a similar illustration. However, this method may be less effective with UMAP, which tends to have more dense islands.
(viSNE maps colored by cell density across three unique conditions - basal, BCR, and IL-7. Arrows highlight the appearance of an abundance difference of a region within the CD4+ T cell compartment. This region is more sparse in the basal condition as compared to BCR and IL-7 conditions.)
Visualize sample heterogeneity across multiple dimensions with DR grid
Many of the views described above are informative when your experiment has a single dimension of interest that you’d like to compare across, like stim condition or disease outcome. For more complex experiments with samples spread across multiple dimensions that you’re interested in comparing, you can use a grid to make easy visual comparisons across these dimensions. For example, in a study with multiple outcomes or stim conditions compared across several time points, you might lay out the outcomes or conditions on the rows, and the time points on the columns (note that depending on your experimental setup, setting up a grid may result in empty plots; just make sure they are expected based on your data, and if something is unexpected, check that your sample tags are correct). Below is an example illustration showing a viSNE grid; you may create a similar illustration for any other DR algorithms by using the appropriate DR channels.
Please refer to the Overview of figure generation using the Illustration Editor for more instructions on how to create the desired layout.
(viSNE grid of stimulation conditions vs individual patient, with various functional markers displayed)
Color DR map by overlay of additional variables
Colored overlay dot plots can help you explore DR results in two different contexts. First, they are useful for comparing traditional manual gating schemes with DR results. Second, they are useful for exploring the relationship between cell populations or groups of similar samples displayed on the DR map and other meta-data variables like disease status, treatment status, or response group. Either of these goals can be achieved using colored overlay dot plots.
Color overlay of manually gated populations
The workflow comparing DR results to manual gates has been demonstrated many times in the literature and can quickly show four important results:
- Populations that were not captured by traditional gates and thus were uncategorized
- Manual gates that are capturing populations that they should not be capturing
- Manual gates that are not capturing all the cells that they should be capturing
- Populations that appear to correlate nicely between the DR map and traditional gates
The workflow is summarized in the visual below where the cells falling into manual gates are colored on the viSNE map:
(viSNE map with cells colored by which manual gate they belong to)
The next image shows an example highlighting the four situations outlined above. The image is a dataset gated manually by a researcher and then subsequently run through viSNE, and all manually gated populations expressed as colors on the viSNE map. Anything colored as dark blue did not fall into a manual gate. Any other color corresponds to a different manual gate:
(situation 1 is seen indicated by a discrete viSNE population with no gate color. Situation 2 (top) is seen by a discrete viSNE population that has the same color as a different discrete viSNE population, showing that a sub-population exists that was not captured by manual gating. Situation 2 (bottom) is also demonstrated by a population that is mostly uncategorized except for small numbers of events captured by various manually gated populations. Situation 3 is shown in the bottom left with a population that seems contiguous in the viSNE map but is only partially captured by manual gates, leading to the spotty coloring. Situation 4 is shown as the small population in the top right that appears categorized nicely both in the viSNE map and by manual gates, since there is little dark blue [ungated] and no other colors simultaneously present)
(Manual gates overlaid on a UMAP map)
Color overlay of other variables
Colored overlay dots can also be used to visualize differences between groups of samples:
(dots colored by their disease tissue type show that the ~800 RNA transcripts used to separate these samples on the viSNE map group them into distinct islands that correlate with their disease type)
Note that the examples shown above describe the overlay of discrete or categorical variables.
Concatenate the DR map across samples
For many of the visualizations described above, it may sometimes be helpful to concatenate the files from multiple samples together. This is often the case if you are trying to compare across groups and have multiple samples per group.
In the Cytobank platform, you can combine events from multiple files into one plot in the Illustration Editor.
An example shown below where the tSNE-CUDA maps of different files are virtually concatenated by stimulation conditions.
(8 files from either the Reference group or BCR-XL stimulated group are concatenated into one plot.)
Please see these articles for more details on the virtual concatenation.
- How to combine events from different files into one plot in the Illustration Editor (Virtual Concatenation)
- How to concatenate dimensionality reduction files for analysis
If you would prefer to physically concatenate your files into one file, please refer to the FCS file concatenation tool for instructions.
Downstream analyses using DR for visualization
Gate the DR map to define populations or groups manually
Once you’ve done some exploratory data analysis by creating different views of your DR map as described above, you will likely want to perform some more quantitative analyses to summarize the data, perform statistical tests, or quantitatively compare DR results to a traditional manual approach. The simplest way to derive quantitative statistics from a DR map is to manually gate the DR map. A simple strategy for manually gating the DR map is to use dot plots colored by Z-axis channel (see above) and the natural separations of the DR map to draw gates. Zooming the gating interface will also help with this process. For regions that are difficult to gate because of a lack of clear separation between more continuous phenotypes, other plot types such as uncolored contour plots and contour plots colored by density can be used to help reveal density trends in the DR map that can guide gate placement. Black dot plots can be used to increase contrast for small populations that might go overlooked in other contexts:
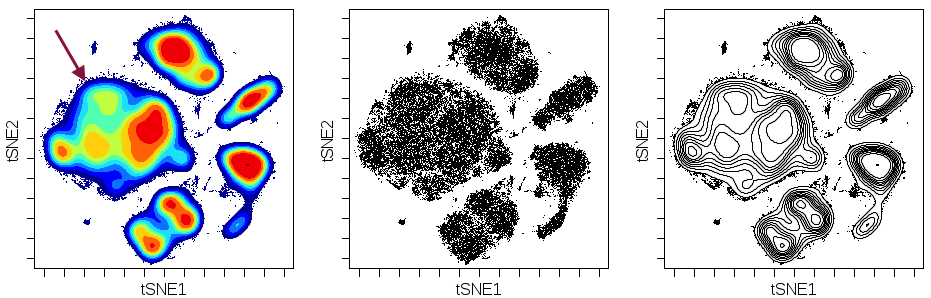
(different plot types such as contour plots colored by density, black dot plots, and uncolored contour plots can help with gating. Density visualizations assist with dissection of phenotypic subsets that are biologically unique but in a more continuous distribution. Arrow indicates one large subset with more continuous smaller nested subsets, as is often seen in T and B cell biology, for example)
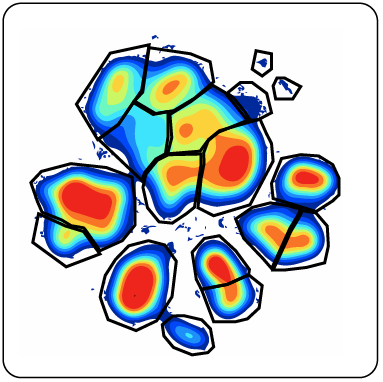
(example of a viSNE map that has been gated)
The simplest workflow for defining groups from your viSNE map or other DR maps is to manually gate the map (as seen above using viSNE as an example). This is a great starting point as you are learning DR algorithms, but it is a somewhat labor-intensive, non-scalable, and subjective process. It can be challenging (just as in traditional sequential gating) to draw gates in places where there are subtle differences between adjacent populations. Furthermore, gates drawn on one DR map aren't portable to future analyses because of the stochastic nature of DR algorithms. Researchers interested in accelerating DR-based workflows should understand options for automated methods for categorizing dimensionality reduction maps.
Create a heatmap of channel expression for all manually gated DR populations
After identifying populations or groups from a DR map, it's often useful to summarize the expression of all channels for each of these populations. This gives you a quick summary of the variety of phenotypes or groups present on a DR map in a condensed, easy-to-interpret space:
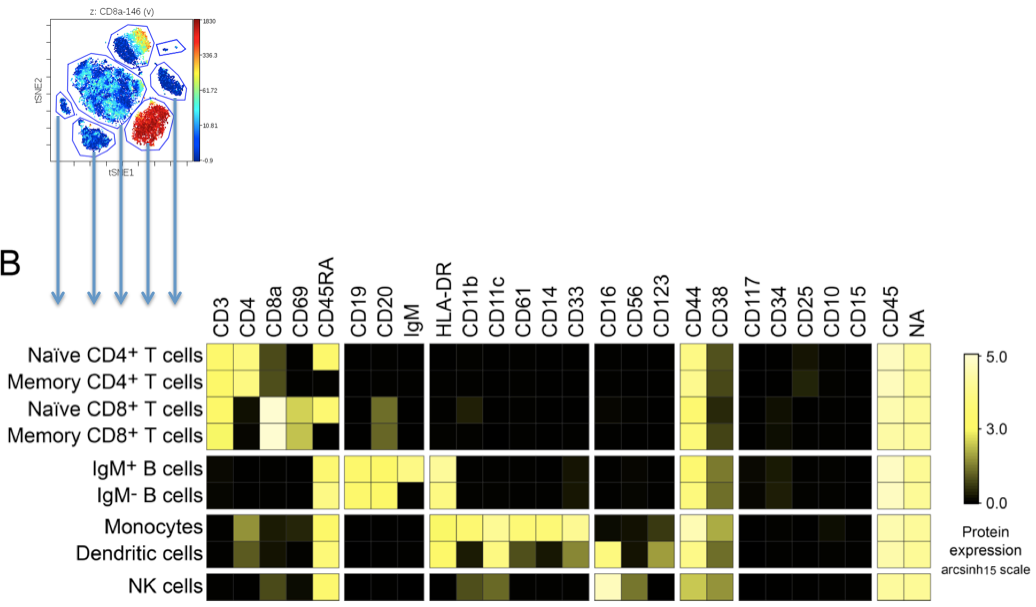
(methodological example of representing populations identified in a viSNE map as a heatmap with their component expression on each channel within the dataset)
Remember that proper scaling of channel values is an important concept when working with heatmaps. Read about how to configure a heatmap of populations versus many channels and how to scale it properly.
Use clustering to automatically define populations or groups and display them on the DR map
A number of methods have been described in the literature whereby cell populations can be defined by semi-automated or automated clustering methods. In Cytobank, you can run SPADE or FlowSOM on the DR generated fcs files, or CITRUS to define cell populations automatically. In addition, computational biologists (or our services team) can use Cytobank API (Application Programming Interface) to run other clustering algorithms that can then be displayed back on the DR map for downstream analyses as described below. The resulting clusters from any of these methods can be displayed back onto the DR map to help visualize the results of quantitative summaries of these clusters.
Generally, you will want to complete your DR analyses before running any method that will cluster your data. This is important, as you will need your files to have the DR channels in the data to be able to overlay clusters onto your DR map. Starting from the DR experiment in Cytobank, run the clustering method using any clustering channels you are interested in (the channels you do not use, including the DR channels, will stay with the data so you can use them for downstream analyses and visualization later). Once the clustering is done, you can follow the cluster gating workflow and then use color overlay dot plots to display the clusters overlaid on the DR axes. Note that overlaying CITRUS clusters specifically on a DR map will require exporting the significant clusters that you want to overlay along with the original files, and then using the FCS files representing the significant CITRUS clusters as the overlaid figure dimension in your color overlay dot plots. When overlaying clusters from any method on a DR map, it may sometimes be helpful to concatenate the files before overlaying the clusters. Please refer to the FCS file concatenation tool for instructions.
(CITRUS identified significant cluster 150101, red, overlaid onto all cells, blue, on the viSNE map)
Just like the manual gating analysis described above, heatmaps and other summaries of clusters can also be created for clusters defined automatically with SPADE and FlowSOM, SPADE on DR, FlowSOM on DR or CITRUS. Additional instructions on creating summaries for these clustering algorithms can be found in their respective support articles.
Have more questions? Submit a request
*For Research Use Only. Not for use in diagnostic procedures.