
Background
A common theme in biomedical research is establishing biological mechanisms to explain observed or suspected differences between multiple populations of individuals. Why do some cancer patients respond well to immunotherapy but others do not? Why does the utility of a particular vaccine diminish among elderly people compared to young people? Why do some people have a slow and painful recovery from surgery but others have a speedy recovery? What are the biomarkers that can help predict these outcomes ahead of time?
High dimensional single cell analysis is an excellent avenue for investigating such questions. Many mechanisms of disease may only be visible at the single cell level, eluding bulk analysis techniques. Emerging technologies such as mass cytometry and improvements to fluorescence flow cytometry are providing unprecedented resolution for observing single cell biology. By analyzing this data, a variety of individual cellular populations can be simultaneously identified, and anomalies in the abundance or signaling behavior of these cellular populations can explain different clinically relevant endpoints. Importantly, they can serve as diagnostics of disease, indications for particular treatment regimens, or predictors of clinical prognosis. The challenge of going from high dimensional data to these useful findings lies in the analysis, which is often manually executed, subjective, and lacking for a variety of reasons. Enter CITRUS.
CITRUS Method Overview
CITRUS (cluster identification, characterization, and regression) is an algorithm designed for the fully automated discovery of statistically significant stratifying biological signatures within single cell datasets containing numerous samples across multiple known endpoints (e.g., responders versus non-responders). CITRUS proceeds through multiple steps, the first of which is unsupervised hierarchical clustering to identify clusters of cellular populations within the overall dataset. After clustered populations are identified, biologically relevant features of these clustered populations are calculated per sample. Features may be either relative abundance of cells in each population compared to the whole sample, or median expression level of functional markers measured across the cells for each population. CITRUS then uses a regularized supervised learning algorithm to determine the populations and features that best predict which endpoint group any given sample belongs to, or uses correlative methods for a correlative measure of the same. The results of a CITRUS run are clusters (populations) that differentiate the observed endpoint of the samples, and the features (relative population abundance or median expression of a functional marker) of the clusters that are responsible.
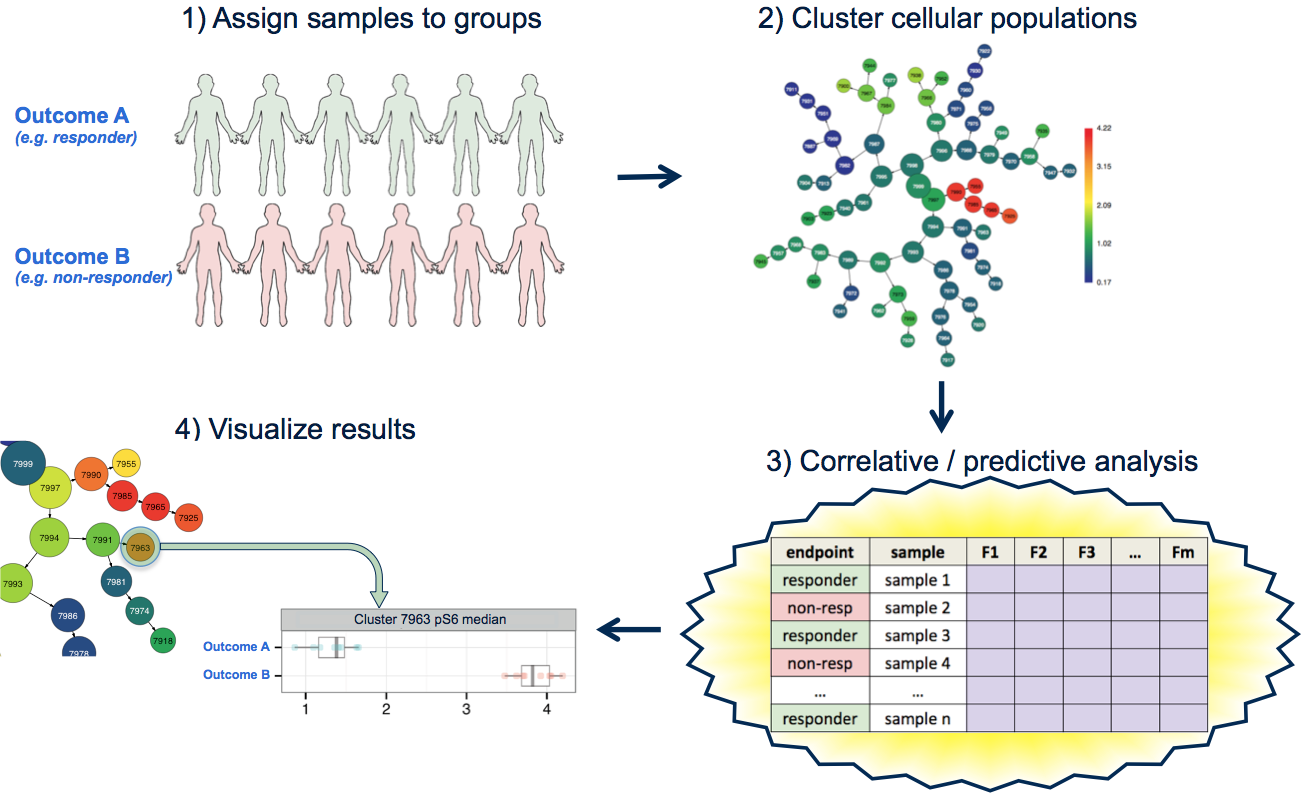
(visual explanation of CITRUS method - in this example a cluster is discovered with differential pS6 signaling in a particular cluster between the responder and non-responder groups)
Getting Started with CITRUS
CITRUS is available on Premium and Enterprise Cytobank (this excludes Community/Basic Cytobank). Learn more about different Cytobank offerings.
Learn how to configure and run a CITRUS analysis.
Applications and Further Reading
CITRUS finds its application in any analysis requiring the comparison of groups of samples as opposed to individual samples. Examples could include clinical comparisons, biomarker discovery, and any application relevant to building a predictive model for the classification of samples in other independent data sets.
Original CITRUS method paper: Bruggner et al, PNAS (2014)
Predicting response to surgery with CITRUS: Gaudillière et al, Science Translational Medicine (2014)
Analyzing differences between immune cells sourced from the liver versus PBMC: Hengst et al, European Journal of Immunology (2015)
The brain’s reward system is causally related to immune response using a mouse model of ventral tegmental area activation: Ben-Shaanan et al. Nature Medicine (2016)
Increased abundance of a unique sub-population of CD4+ T cells predicts increased tolerance to liver transplant (without immunosuppression) in children: Lau et al. Pediatric Transplantation (2016)
Read more about the power of CITRUS and how to harness it through support articles on CITRUS or through a blog series on getting the most out of CITRUS.
Video Overview of CITRUS
Also make sure to check out our articles on how to setup a CITRUS analysis or how to analyze CITRUS results.