
Background
CytobankBridgeR (AKA "BridgeR") is a package written for R that contains various tools to interact with Cytobank. This package leverages the Cytobank API in order to extend and support native Cytobank functionality in workflows that are easy to follow for users who are unfamiliar with R. It does this using scripts and pipelines, that leverage the CytobankAPI R package and other R functions and packages. Click the links below to jump to any section in this article. Click the links below to jump to any section in this article.
Pre-requisites
Before being able to utilize the CytobankBridgeR package, ensure you have installed the required tools necessary:
- A working version of R (RStudio is also recommended)
- The CytobankAPI and CytobankBridgeR package
Follow the Using R to Interface with Cytobank Prerequisites Guide to walk through these necessary requirements step by step.
Workflows
Familiarizing yourself with the CytobankAPI R package will help, but is not necessary to follow any of these guides.
Learn more about the CytobankAPI package.
Apply Cluster Gates
Background
Apply cluster gates is a tool to help define groups (gates) around individual integers or sets of integers in a “cluster channel.” Cluster channels are columns in your data that contain integers that assign events or rows in the data into groups. There are two types of groups assigned by cluster channels:
- Cluster groups: Cluster channels can contain integers that assign the data into groups based on the results of a clustering algorithm (such as SPADE) that analyzes raw data across a number of parameters.
- Annotation groups: Cluster channels can contain integers that assign the data into groups based on some other categorical variables that might be, for example, overlayed on top of raw data to help visualize correlations between the variables. Annotation channels are often used to code variables such as gender, disease type, treatment, etc in bulk data.
Gates are used to define groups from cluster channels to stratify the data in downstream analyses or visualizations. Learn more about clustering channels and clustering gates.
How to use
1. Choose whether you would like to define one group per individual integer value, or combine sets of integers from a cluster channel into a single group.
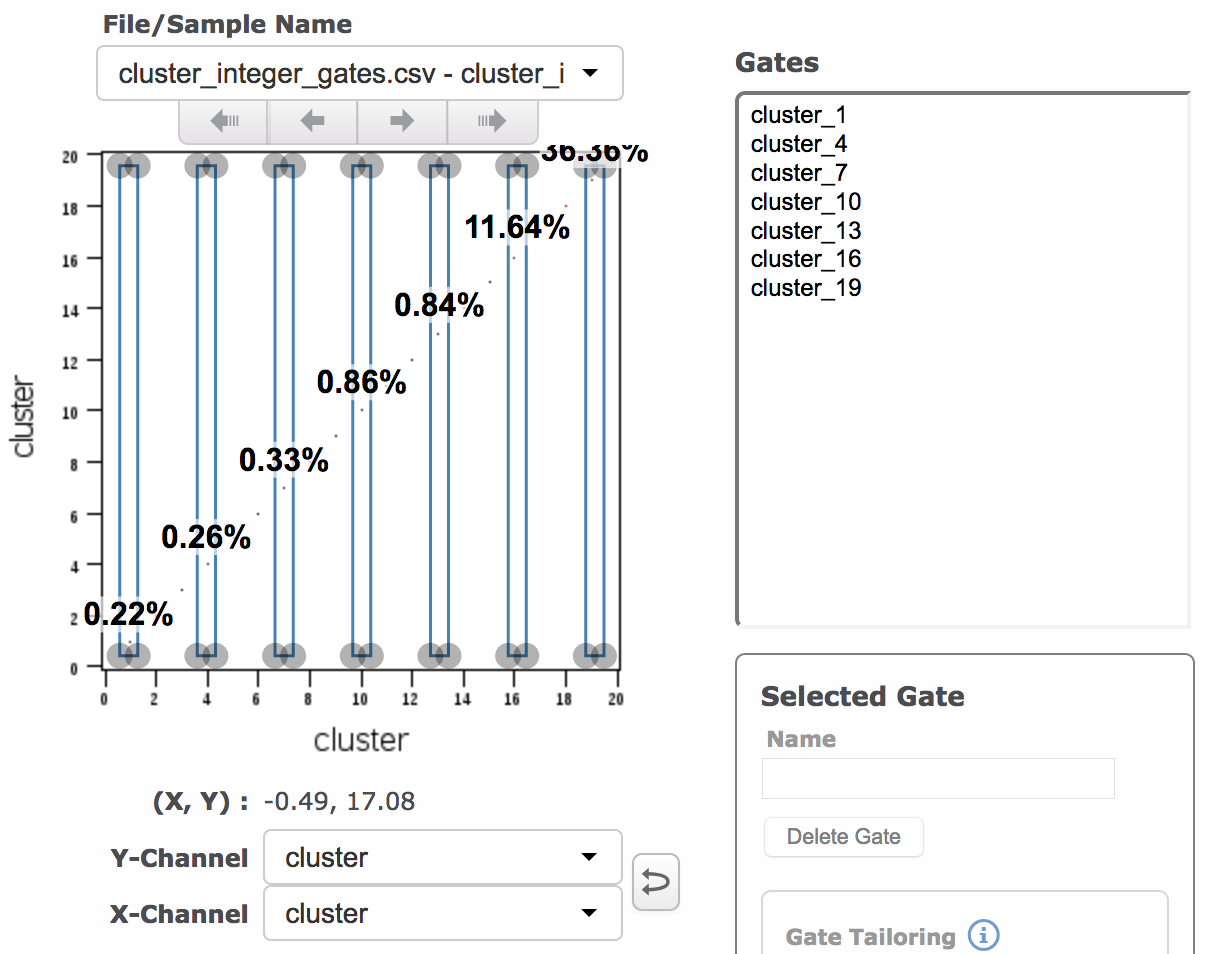
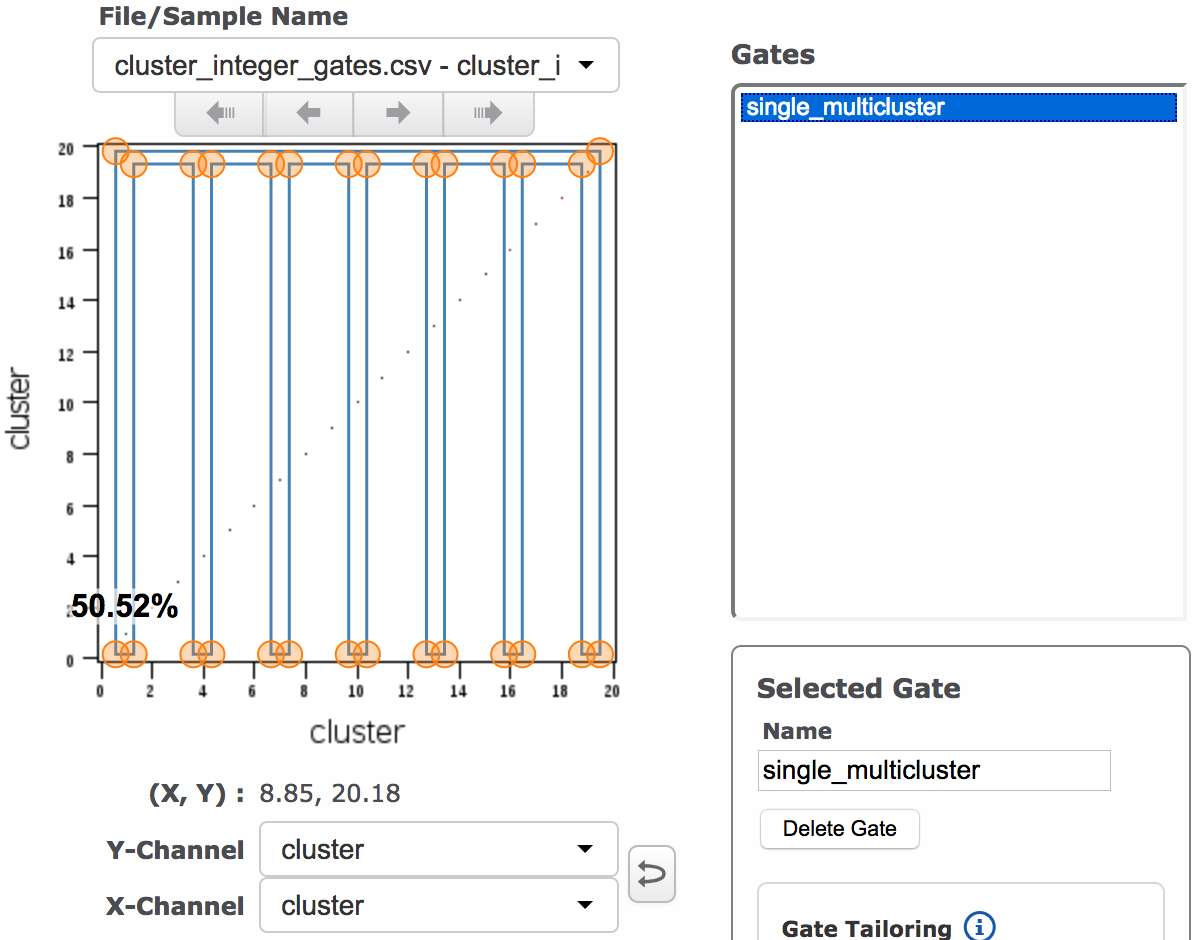
(Left: many gates, each containing an individual integer. Right: one gate that combines multiple integer values; 1, 4, 7, 10, 13, 16 and 19 are all included in the gate.)
- If you would like to define one group per individual integer value, you will set the grouped parameter below to FALSE.
- If you would like to combine sets of integers into a single group (like an OR gate), you will set the grouped parameter below to TRUE and run the apply cluster gates function once for each group you want to define.
2. Generate an API token from your Cytobank. Copy the generated token.
3. Authenticate and create a Cytobank Session via the ‘authenticate’ function.
- Copy and paste the line of code below into your console in R or RStudio
cyto_session <- CytobankAPI::authenticate(site="premium", user="cytobank_user", auth_token="CYTOBANK_AUTH_TOKEN")
- Replace CYTOBANK_AUTH_TOKEN (leave the quotation marks around it) with your API token
- Replace premium with the name of your Cytobank server (if it’s not premium)
- Replace cytobank_user with your Cytobank username
- Hit enter to run the code
![]()
4. Once authenticated, utilize the CytobankBridgeR::gates.apply_cluster_gates function:
- Copy and paste the code below into your console
CytobankBridgeR::gates.apply_cluster_gates(UserSession=cyto_session, experiment_id=EXPERIMENT_ID, clusters=c(1, 2, 3, 4), name="CLUSTER_NAME", channel_name="CHANNEL_NAME", grouped = FALSE)
- experiment_id: Find the Experiment ID from the Cytobank experiment where you want to apply these gates and paste it into the code to replace EXPERIMENT_ID.
- clusters: Replace clusters with the integers representing the clusters you want to create groups for. By default, each value will be contained within its own cluster group (grouped = FALSE). To combine sets of integers into a single group, read the section pertaining to the grouped parameter below.
- Pro tip: If you want to create groups for a long list of integers, you can list only the first and last integers separated by a colon. For example, if you want to create groups for every integer from 1 to 200, you could replace 1, 2, 3, 4 with 1:200.
- name: Replace CLUSTER_NAME with the prefix you’d like to use for each cluster gate.
- i.e. if clusters=c(1,2,3) and name=”cluster_”, the populations created will be named cluster_1, cluster_2, and cluster_3 respectively.
- channel_name: Replace CHANNEL_NAME with the name of the short channel to cluster on (learn more about short channel and long channel names). This channel should contain integers that assign events or rows in the data into groups and will be utilized as both the X & Y axis for cluster gating.
- grouped: If you chose to combine sets of integers into a single group, change the grouped parameter to TRUE. You will need to run the function once for every combined integer gate you want to create. There are two parameters you will need to change each time:
- In this case, clusters will contain the integers that you want to include in a single group.
- The name parameter will be the name for the single gate you create each time you run the function.
- Hit enter to run the code

(Multiple individual integer clusters) (Single multiple integer cluster)
Advanced Settings
If the above instructions are not providing visible plots or gates, the most likely issue is scale arguments. By default, each time you run the gates.apply_cluster_gates function, the scales are set from 0 to 1 + the maximum value given in the clusters parameter. The min and max of the scale can instead be set by utilizing the integer_min and integer_max parameters to set scales manually. In addition, if the function is not properly applying cluster gates due to timeout issues, set the timeout parameter to a larger value.
integer_min: integer representing the minimum integer in the cluster channel [optional]
- The default is set to 1, this assumes clusters begin at 1, but can be changed if there is any +/- offset
- The minimum scale will be set to integer_min-1 (0 by)
integer_max: integer representing the maximum integer in the cluster channel [optional]
- The default is the maximum number presented within the clusters vector, but can be set to any integer
- The max scale will be set to integer_max+1
timeout: integer representing the request timeout time in seconds [optional]
Bubble SPADE Nodes
Purpose
Bubbling all SPADE nodes individually is useful for workflows that involve exporting individual SPADE node data for analysis without “bubbling” (grouping) them. Workflows where this might be useful include:
- Running SPADE on viSNE map coordinates (read more about SPADE on viSNE here).
- Exporting SPADE nodes for automated or semi-automated analyses to help group the nodes
Background
Bubble spade nodes is a tool to help bubble individual SPADE nodes into their own singular group that can be later exported for analysis.

How to use
1. Generate an API token from your Cytobank. Copy the generated token.
2. Authenticate and create a Cytobank Session via the ‘authenticate’ function.
- Copy and paste the line of code below into your console in R or RStudio
cyto_session <- CytobankAPI::authenticate(site="premium", user="cytobank_user", auth_token="CYTOBANK_AUTH_TOKEN")
- Replace CYTOBANK_AUTH_TOKEN (leave the quotation marks around it) with your API token
- Replace premium with the name of your Cytobank server (if it’s not premium)
- Replace cytobank_user with your Cytobank username
- Hit enter to run the code
![]()
3. Generate a SPADE object in R
In order to use this tool, a SPADE object in R must be generated first. Generating a SPADE object requires knowing the SPADE ID of the specific SPADE analysis you want to bubble nodes for. This is retrievable by looking at the URL for this specific SPADE analysis. Find out more about finding feature IDs within Cytobank.
- Copy and paste the code below into your console
spade <- CytobankAPI::spade.show(UserSession=cyto_session, experiment_id=EXPERIMENT_ID, spade_id=SPADE_ID)
- experiment_id: Find the Experiment ID from the Cytobank experiment where you want to apply these SPADE bubbles and paste it into the code to replace EXPERIMENT_ID.
- In the above example URL, the Experiment ID is 1234.
- In the above example URL, the Experiment ID is 1234.
- spade_id: Find the SPADE ID from the Cytobank SPADE analysis where you want to apply these SPADE bubbles and paste it into the code to replace SPADE_ID.
- In the above example URL, the SPADE ID is 5678.
![]()
4. Utilize the CytobankBridgeR::spade.bubble_spade_nodes function:
- Copy and paste the code below into your console
CytobankBridgeR::spade.bubble_spade_nodes(UserSession=cyto_session, spade=spade)
- Hit enter to run the code
![]()
(Looking at the corresponding SPADE tree in the Cytobank app, you will see individual SPADE nodes bubbled)
Advanced Settings
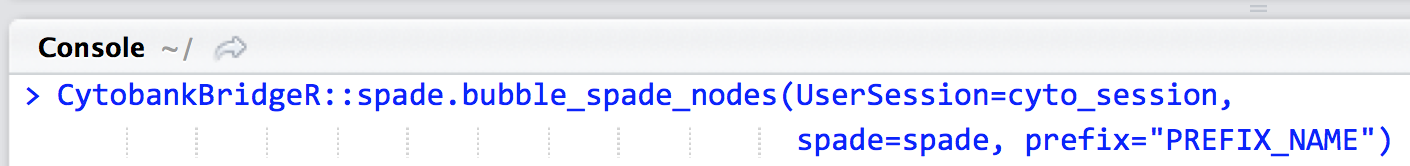
If you’d like to set a custom prefix to name the bubbles, utilize the prefix argument. The default prefix is “cluster_” so that, for example, each bubble in a three-node tree would be named cluster_1, cluster_2 and cluster_3.
- Copy and paste the code below into your console
CytobankBridgeR::spade.bubble_spade_nodes(UserSession=cyto_session, spade=spade, prefix="PREFIX_NAME")
- prefix: Replace PREFIX_NAME with the prefix you’d like to use for each bubble.
- i.e. if prefix="cluster_", the bubbles created will be named cluster_1, cluster_2, and cluster_3, …, cluster_n respectively.
Stable Download of FCS Files
Purpose
The most stable form of downloading FCS files from Cytobank is to download them one by one. This ensures maximum stability because each file will be downloaded before moving onto the next. Using this tool provides an efficient and effective method for downloading FCS files.
Background
Stable download of FCS files is a tool to help download FCS files from Cytobank in the most stable manner. There are some extra mechanisms involved that help maximize the stability and performance of the downloads, including compressing the FCS file by zip downloading, and then extracting the FCS file afterwards.
How to use
1. Generate an API token from your Cytobank. Copy the generated token.
2. Authenticate and create a Cytobank Session via the ‘authenticate’ function.
- Copy and paste the line of code below into your console in R or RStudio
cyto_session <- CytobankAPI::authenticate(site="premium", user="cytobank_user", auth_token="CYTOBANK_AUTH_TOKEN")
- Replace CYTOBANK_AUTH_TOKEN (leave the quotation marks around it) with your API token
- Replace premium with the name of your Cytobank server (if it’s not premium)
- Replace cytobank_user with your Cytobank username
- Hit enter to run the code
![]()
3. Get a list of possible FCS files to download
In order to use this tool, find the Experiment ID by looking at the URL for the specific experiment that you want to download FCS files from.
When stable downloading FCS files via CytobankBridgeR, a vector of FCS file IDs must be retrieved. Utilize the CytobankAPI::fcs_files.list function to gather the required information:
- Copy and paste the code below into your console
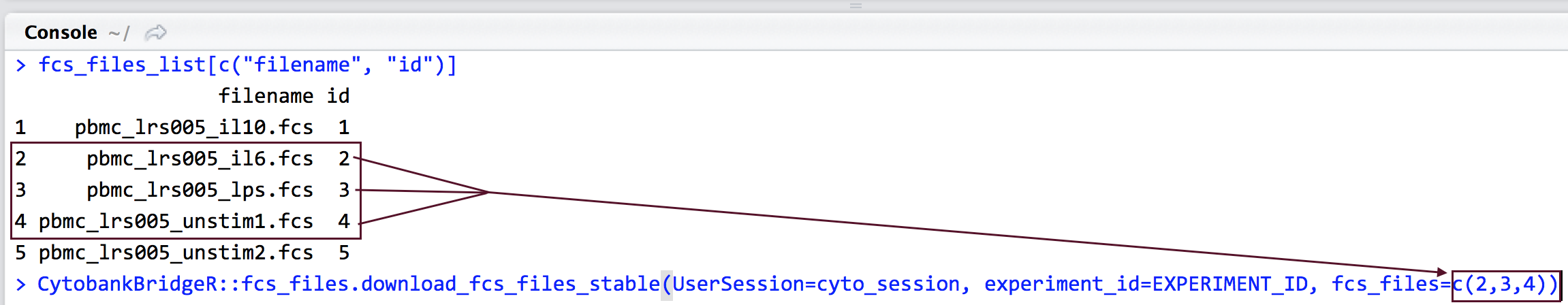
fcs_files_list <- CytobankAPI::fcs_files.list(UserSession=cyto_session, experiment_id=EXPERIMENT_ID); fcs_files_list[c("filename", "id")]
- experiment_id: Find the Experiment ID from the Cytobank experiment where you want to download FCS files from, and paste it into the code to replace EXPERIMENT_ID.
- In the above example URL, the Experiment ID is 1234.
- Hit enter to run the code

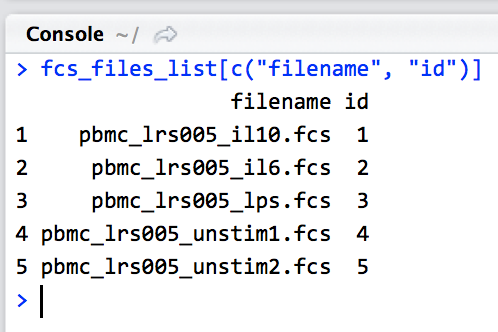
From the resulting list of FCS files available in the experiment, select the ones you want to download and make a list of their id numbers.
4. Utilize the CytobankBridgeR::fcs_files.download_fcs_files_stable function:
- Copy and paste the code below into your console
CytobankBridgeR::fcs_files.download_fcs_files_stable(UserSession=cyto_session, experiment_id=EXPERIMENT_ID, fcs_files=c(2,3,4))
- experiment_id: Paste the experiment ID from Cytobank that you identified above in step 3 into the code to replace EXPERIMENT_ID.
- fcs_files: Replace fcs_files with a comma-separated vector of the id numbers that you identified in step 3 representing the FCS files you want to download.
- Pro tip: If you want to download FCS files for a long list of FCS file IDs, you can list only the first and last FCS file ID separated by a colon. For example, if you want to download FCS files for every FCS file with an ID from 1 to 200, you could replace 2, 3, 4 with 1:200.
- Note: if not all files show, try setting max.print to a larger value (default=1000)
- i.e. options(max.print=5000)
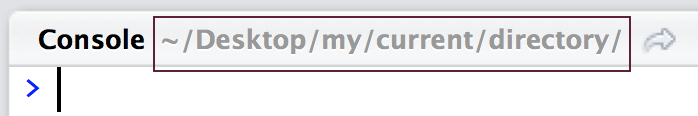
Files are downloaded by default to the current directory in which R currently is in. This is shown in the upper left corner of the console.
- The folder in which the FCS files will be downloaded to will be of the following structure:
- CytobankExperimentID will be the Experiment ID unique to your Cytobank
- experiment_CytobankExperimentID_fcs_files
Advanced Settings
Below, there are options that show particular advanced settings that may be helpful when downloading FCS files via this stable method. This includes setting custom download directories, changing download protocols, and extending the timeout of a particular FCS file if it fails to download within the default 5 minutes per FCS file.
CytobankBridgeR::fcs_files.download_fcs_files_stable(UserSession, experiment_id, fcs_files, directory=getwd(), zip_download=TRUE, timeout=300)
directory: To choose a different directory to download the FCS files to, enter a specific file directory separated by "/" (forward slash)
- I.e. directory="/new/file/directory"
- By default, the directory selected will be the one currently shown in the console
zip_download: To change the way in which FCS files are downloaded, choose TRUE [default] or FALSE to choose whether or not to download each file one by one via ZIP download [default] or normal FCS download [optional]
timeout: If particular files are taking over 5 minutes to upload [default time], increase this value to something larger. This is represented in seconds [default=300]